Sindrome di Drave: cause, sintomi, diagnosi e trattamento
La sindrome di Drave è una forma resistente (non trattabile) di epilessia associata a convulsioni indotte da febbre. Il tipo di ereditarietà autosomica dominante è caratteristico.
È una grave epilessia mioclonica infantile (TMEM). Questa patologia è anche chiamata encefalopatia nei neonati. Si verifica con una frequenza di 1: 40000 neonati, i ragazzi sono malati 2 volte più spesso. Fa il suo debutto nel primo anno di vita e manifesta febbre (con un aumento della temperatura) e afezioni afezioni (a temperatura normale) generalizzate e locali con la presenza di contrazioni muscolari a breve termine (mioclonia). I bambini sono in ritardo nello sviluppo mentale, non sono suscettibili al trattamento antiepilettico.
classificazione
Si riferisce a forme di epilessia che hanno sintomi di epilessia sia generalizzata che parziale.
Sfondo storico
La sindrome fu scoperta nel 1978 dall'epilettologo Charlotte Drave e prende il nome dall'autore. Fino a quel momento, questa forma di epilessia era attribuita alla sindrome di Lennox-Gastaut.
Molti autori considerano il nome TMEM non riuscito, dal momento che le crisi miocloniche di solito si uniscono diversi anni dopo il debutto, e in alcuni casi possono essere assenti.
Nel 1999, la Germania ha proposto il termine "grave epilessia idiopatica con segni di grand mal in tenera età".
Nel 2001, il Giappone: l'infanzia hemi-grand mal.
Nel 2004, in Francia: epilessia polimorfica dell'infanzia.
Termine eponimicheskom attualmente usato - Sindrome di Drave.
eziologia
È la patologia dei canali del sodio. Normalmente, i canali del sodio mantengono una quantità costante di sodio nella cellula e la sua attività elettrica. La sindrome di Drave si sviluppa in individui con un difetto genetico nei canali del sodio. Questo fenomeno fu scoperto nel 2001 quando fu isolata una mutazione del gene SCN1A (una subunità del canale del sodio). Dal 2001, gli studi sulla mutazione genetica sono continuati. Attualmente sono noti più di 100 tipi di danno genetico SCN1A.
Ad

Le mutazioni sono diverse e possono aumentare, diminuire o non modificare l'eccitabilità delle cellule nervose. La sindrome di Drave si verifica solo nel caso di aumentata eccitabilità dei neuroni ed è l'unica sindrome epilettica, la cui diagnosi si basa sull'analisi del DNA.
Nel 20% dei casi, la mutazione genetica non è la causa della malattia, l'eziologia non è nota. Ci sono prove di una storia familiare di convulsioni febbrili ed epilessia.
Quadro clinico
prima sintomi della sindrome La Drava può presentarsi all'età di 7 anni. Il corso della malattia è in scena.
- Debutto sul palcoscenico Si alza all'età di 1 anno. Manifestato sotto forma di convulsioni febbrili Si verificano con le figure a bassa temperatura e sono di natura breve. La trasformazione delle convulsioni febbrili in varie forme di epilessia varia dal 3 al 5%.
- Fase "catastrofica". All'età di 1-4 anni le convulsioni febbrili possono essere sostituite da afebrile e diventare epilettiche.
- Fase di stabilizzazione. Dopo 4-5 anni, le convulsioni si verificano meno frequentemente? e il deterioramento cognitivo lordo (perdita di memoria, deficit intellettivo) e deficit neurologico (encefalopatia epilettica) vengono alla ribalta.
Stato neurologico
Prima delle manifestazioni cliniche, i bambini sono sani, lo sviluppo fisico e neuropsichiatrico soddisfa gli standard di età. Dopo il debutto, appare una diminuzione del tono muscolare, aumentano i riflessi tendinei, si sviluppano segni di insufficienza piramidale. Dopo 1 anno, i bambini hanno un'andatura atossica (traballante) e movimenti goffi.
Disturbi extrapiramidali sotto forma di aumento del tono muscolare, tremore e rigidità dei movimenti si uniscono all'età scolare. il pubertà la sintomatologia è completata da tremore intenzionale (insorge durante il movimento ed è assente a riposo) e mioclono non epilettico (contrazione spontanea e rilassamento di un'area muscolare). La malattia è complicata da deformità scheletriche (cifoscoliosi, piede piatto).
Ad
La progressione dei disturbi neurologici continua fino a 8 anni dopo, la condizione si stabilizza.
Sviluppo cognitivo (tutti i tipi di attività mentale)
All'inizio della malattia, lo sviluppo psicoverbale rallenta e in seguito arriva la sua completa regressione. In parallelo, ci sono disturbi comportamentali (iperattività, autismo). All'età di 5 anni si sviluppa un ritardo mentale. Riducendo la frequenza degli attacchi, è possibile migliorare le funzioni cognitive.
Caratteristiche degli attacchi
- Assansia atipica. Trovato nel 40-95% dei bambini. L'attacco si sviluppa gradualmente sullo sfondo delle normali attività. Nasce dall'età di 4 mesi a 6 anni. È caratterizzato da un atomo (omissione delle spalle, un cenno passivo, un'improvvisa caduta) o una componente mioclonica (cenni ritmici attivi, mioclono delle palpebre e dei muscoli della cintura della spalla). Accompagnato da una pronunciata colorazione vegetativa (decolorazione della pelle, aumento del lavoro delle ghiandole salivari e sudoripare), disturbi dell'andatura. Durata da diverse ore a diversi giorni. I sintomi sono più pronunciati dopo il risveglio.
- Convulsioni epilettiche massive (mioclono). È il principale sintomo clinico e si sviluppa un anno o più dopo il debutto all'età di 1,5-4 anni. A poco a poco, l'intensità e la frequenza degli attacchi aumenta. Il mioclono è più comunemente visto nei muscoli del collo e della schiena. Viene mostrato da "annuire", scuotendo le spalle con "alzando" le braccia, inclinando ritmicamente la testa indietro, piegando il corpo. Per gli attacchi caratterizzati da serialità, l'aumento al risveglio e l'addormentarsi, l'alta sensibilità alla luce.
- Mioclono non epilettico. Si sviluppa dopo 4-5 anni. È provocato dai movimenti. Copre le sezioni finali degli arti e ha un'ampiezza bassa. Le contrazioni muscolari sono visivamente difficili da notare, ma puoi palpare. Non ci sono cambiamenti sull'EEG.
- Emoconvulsioni alternate. Si verificano su uno o sull'altro lato del corpo.
- Attacchi focali Trovato nel 43-79% dei bambini. Ci sono motori complessi, automotori, parossismi versi.

Diagnosi della sindrome di Drave
Sviluppo graduale e graduale del quadro clinico e debutto con convulsioni febbrili. Ci vogliono 1-2 anni dall'esordio delle manifestazioni all'instaurarsi di una diagnosi La diagnosi è fuori dubbio in una tipica clinica della malattia. È possibile sospettare la sindrome di Drave in qualsiasi bambino normalmente in fase di sviluppo con convulsioni febbrili prolungate iniziate nel primo anno di vita e che si sono sviluppate dopo crisi epilettiche di qualsiasi tipo e disturbo dello sviluppo.
Ad
La diagnosi si basa su dati di esami clinici, analisi di convulsioni, modifiche EEG e osservazione a lungo termine del paziente.
Criteri di diagnosi:
- A partire da 1 anno di vita.
- Convulsioni febbrili
- Convulsioni miocloniche dopo 2 anni.
- Assensia atipica, focale, emiciclivulsiva e emiconvulsioni alternate.
- Alta frequenza di convulsioni, stato epilettico.
- Violazioni pronunciate dell'attività mentale.
- EEG. C'è uno sviluppo graduale delle variazioni EEG. Nel primo anno di vita, gli indicatori corrispondono alla norma, nel secondo anno di vita rivelano un rallentamento dell'attività principale, una combinazione di attività epilettiforme regionale e diffusa, manifestazioni di sensibilità alla luce. Attualmente, il monitoraggio del sonno EEG video sta diventando sempre più diagnostico.
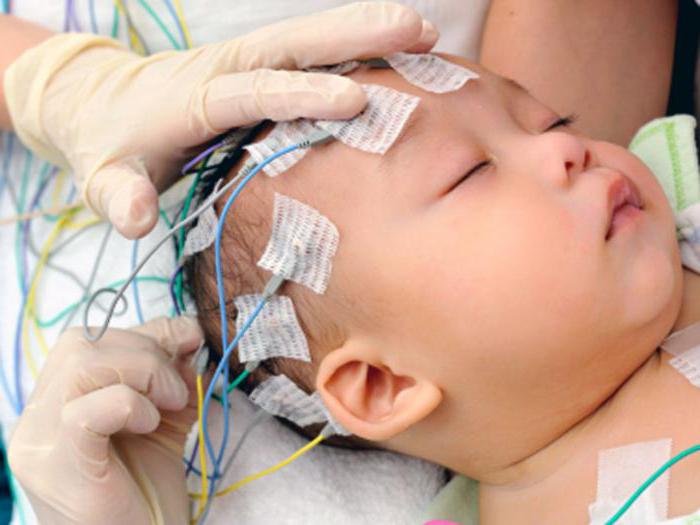
- Neuroimaging con risonanza magnetica: atrofia diffusa della corteccia cerebrale, che è più pronunciata nei lobi frontali e temporali; atrofia cerebellare; aumento moderato ventricoli del cervello. Sono descritti casi singoli sotto forma di cambiamenti locali: cisti, sclerosi temporale, riduzione del volume del lobo temporale, dislessinazione.
- Analisi del DNA: rilevazione di una mutazione nel gene SCN1A, che può essere assente nella sindrome di Drave.
- Modalità di ereditarietà autosomica dominante. Se c'è la possibilità di consultare la genetica.
Trattamento per la sindrome di Drave
Il TMEM è uno dei più resistenti al trattamento delle sindromi. Non ci sono anticonvulsivanti altamente efficaci per il controllo delle convulsioni nella sindrome di Drava. In tenera età, barbiturici e benzodiazepine possono essere efficaci per un breve periodo; in età avanzata, la combinazione di valproato e topiramato è ottimale.
Farmaci antiepilettici con un effetto positivo:
- La droga di scelta - "Valproat". Aumenta il contenuto di GABA nel sistema nervoso centrale.
- Benzodiazepine. Riduce l'eccitabilità dei neuroni grazie all'affinità con il neurotrasmettitore inibitorio GABA.
- Barbiturici. Inibisce la trasmissione degli impulsi nervosi.
- "Etosuccimide." Inibisce la distruzione di GABA, riduce la conduttività degli impulsi nervosi.
- "Topiramato". Aumenta l'attività di GABA.
Farmaci antiepilettici che possono aggravare l'attacco:
Ad
- "Lamotrigina".
- "Fenitoina".
- "Carbamazepina."
- "Vigabatrin".
Trattamento con corticosteroidi, immunoglobuline, dieta chetogenica e stimolazione nervo vago non ha mostrato efficienza.
È molto importante monitorare la temperatura corporea, specialmente nel periodo di una malattia infettiva. Questi bambini hanno dimostrato di ridurre la temperatura sopra i 37 gradi prendendo antipiretici. Il monitoraggio della temperatura deve essere effettuato al mattino e alla sera. È necessario escludere il surriscaldamento del bambino (bagni caldi, esposizione al sole). I genitori devono conoscere la fotostimolazione (esposizione alla luce). Alcuni bambini possiedono l'autostimolazione e ne traggono piacere. Causano un attacco coprendo gli occhi e sollevando gli occhi verso l'alto con una contrazione ritmica delle palpebre. Questo gruppo di bambini sta vivendo ossessioni ricevendo un tale "piacere" dubbioso e con il sollievo medico degli attacchi, non lasciano tentativi di provocare di nuovo un attacco.

prospettiva
La remissione è estremamente difficile da raggiungere. L'obiettivo del trattamento è ridurre il numero di crisi, prevenire episodi di serialità e lo sviluppo dello stato epilettico. È necessario migliorare gli indicatori delle funzioni cognitive, adattare i bambini nella società. All'età di 12-14 anni, gli attacchi diventano meno frequenti e appaiono i disturbi vegetativi e astenici e i disturbi mentali. La mortalità nell'infanzia è del 20%.