Fibrosi cistica: sintomi, trattamento, diagnosi, segni della malattia
Ogni persona è felice e soddisfatta quando il suo corpo funziona correttamente, non si verificano sensazioni dolorose negli organi interni e lo stato di salute non lascia molto a desiderare.
Tuttavia, l'impatto di vari fattori, che vanno dalla predisposizione genetica all'impatto negativo dell'ambiente naturale, porta alla comparsa di patologie che si manifestano sotto forma di un insieme di segni esterni.
Una delle malattie più gravi trasmesse per eredità è considerata la fibrosi cistica, i cui sintomi sono molto diversi. È associato a disordini genetici nelle cellule umane e non può essere curato. Gli organi respiratori, il tubo digerente e alcuni altri sistemi sono principalmente colpiti. La fibrosi cistica è insolitamente acuta nei bambini.
Ad
Comprenderemo la questione in modo più dettagliato.
Concetti di base
La fibrosi cistica, di cui parleremo un po 'più tardi, ha anche un altro nome: la fibrosi cistica del pancreas. Questo termine è ampiamente usato nella scienza medica dei paesi del Nuovo Mondo (USA, Canada) e Australia. In Europa, come nel nostro paese, è comune chiamare questa patologia chiamata fibrosi cistica. Il trattamento, ovviamente, non dipende dalla terminologia, ma si basa sui dati ottenuti durante l'esame. 
Non c'è bisogno di pensare che questa malattia sia rara. La fibrosi cistica nei bambini viene diagnosticata in almeno quattro casi ogni 10.000 bambini. Colpisce i bambini, sia maschi che femmine.
La patologia, come già accennato, è ereditaria, cioè è impossibile prenderla. Non è sempre possibile, subito dopo la nascita, stabilire che il neonato ha fibrosi cistica. I sintomi della malattia potrebbero non apparire per un periodo piuttosto lungo.
Ad
La medicina ha conosciuto casi in cui il disturbo è stato diagnosticato in pazienti che hanno raggiunto la maturità. Tuttavia, nella stragrande maggioranza dei casi, stiamo parlando dei primi mesi o anni di vita, cioè la fibrosi cistica si trova più spesso nei neonati.
Caratteristiche del meccanismo di trasmissione della malattia
Un disturbo genetico può essere ottenuto solo nel caso in cui il bambino erediti il gene modificato sia dalla madre che dal padre. Altrimenti, il paziente è considerato solo il portatore della malattia. Il test della fibrosi cistica non darà risultati positivi.
Il fatto è che la struttura genetica di ogni persona è accoppiato, cioè un bambino riceve un gene da ciascuno dei genitori. Alcuni dei geni ottenuti non sono in grado di funzionare normalmente a causa di eventuali danni.
Ma per la fibrosi cistica nei bambini che hanno ricevuto geni alterati da sviluppare completamente, entrambi sono necessari. Questo meccanismo, a proposito, è inerente alla stragrande maggioranza delle patologie genetiche.
Quindi, se entrambi i genitori sono portatori, il bambino può ammalarsi con una probabilità del 25%.
Tutte le forme di fibrosi cistica si manifestano nei bambini prima che raggiungano i 2 anni di età. Solo nel 30% dei casi i segni esterni rimangono nascosti per un periodo più lungo. Recentemente, tuttavia, i nuovi metodi diagnostici, come lo screening neonatale, stanno diventando più comuni. Ti permette di identificare i segni della malattia in età molto precoce.
Il progresso della patologia
La patogenesi della malattia non è completamente determinata da nessun medico specialista. La fibrosi cistica negli adulti e nei bambini inizia con il rilascio di una sostanza specifica da parte di alcune ghiandole interne del corpo, che ha una densità e una viscosità molto elevate.
Questo segreto interno, grazie alle sue proprietà fisiche, intasa la maggior parte dei dotti nelle mucose degli organi interni. Come risultato del processo in questione, ci sono:
- fibrosi cistica;
- forma intestinale di fibrosi cistica;
- fibrosi cistica delle ghiandole mucose e altri organi simili.
Lo sviluppo della malattia porta alla comparsa di processi secondari nei polmoni, nel fegato, nel pancreas e nel tratto gastrointestinale. Ciò è dovuto principalmente alle violazioni nel processo di secrezione degli enzimi, nonché alla comparsa di focolai di infiammazione, che influiscono negativamente sul corretto funzionamento degli organi presi in considerazione.

La fibrosi cistica, i cui sintomi possono manifestarsi, come già accennato, in molte ghiandole del corpo, porta a una mancanza di fosforo, potassio e sodio nel corpo. A proposito, molti esperti ritengono che questo sia il motivo per cui i segreti nascosti hanno un'alta viscosità.
Complicazioni negli organi interni
Nel corpo di una persona a cui è stata diagnosticata la fibrosi cistica, viene secreto un fluido denso secretivo. Come risultato dell'ostruzione dei dotti in alcuni organi e sistemi del corpo, inizia il processo di formazione di tumori di piccole dimensioni (le cosiddette cisti).
Il muco che rimane all'interno della persona agli stadi avanzati del decorso della malattia causa atrofia dei tessuti delle ghiandole endocrine, oltre a sviluppare la fibrosi. Quest'ultimo, in particolare, porta alla graduale sostituzione dei tessuti normali degli organi endocrini con cellule connettive.
I primi cambiamenti sclerotici negli organi sono spesso accompagnati da fibrosi cistica. La diagnosi di tali cambiamenti è ostacolata dal fatto che il processo è accompagnato dall'aspetto e dalla crescita dell'infiammazione purulenta.
Disfunzione nei polmoni
La malattia porta al fatto che il sistema bronchiale nei polmoni cessa di funzionare correttamente non a causa di difficoltà nel processo di scarico del muco del muco (a causa della maggiore viscosità del segreto), ma a causa di processi infiammatori cronici che si verificano in questo organo.
La disfunzione dell'apparato respiratorio, in particolare i polmoni, inizia con la comparsa dell'ostruzione dei bronchioli nei bronchi. La presenza in questi organi interni della massa mucopurulenta porta al loro aumento di dimensioni e, di conseguenza, al blocco degli spazi tra i bronchi. Alla fine, il paziente con fibrosi cistica inizia a sperimentare difficoltà di respirazione a causa del blocco completo di ampie aree dei polmoni, cioè l'otturazione.
La flemma che si accumula nei polmoni, oltre ai problemi con la respirazione, provoca un alterato afflusso di sangue all'organo. Esternamente, questo si manifesta sotto forma di una tosse forte e dolorosa, che accompagna sempre la fibrosi cistica, i cui sintomi sono ora considerati.
Il deterioramento del sistema immunitario porta al fatto che il sistema respiratorio umano è esposto all'infezione da una serie di altri microrganismi, ad esempio lo stafilococco o il bacillo di Pseudomonas. Ciò è dovuto a:
- una diminuzione del livello di interferone;
- una diminuzione del numero di anticorpi;
- diminuita attività fagocitaria.
Gli agenti patogeni infettivi continuano le azioni distruttive nel corpo del paziente, che distrugge lo strato epiteliale interno dei bronchi.
Malattie concomitanti dell'apparato respiratorio
La fibrosi cistica nei bambini e negli adulti porta a una varietà di patologie che, a prima vista, non sono associate alla malattia genetica in esame.
In particolare, si verificano polmonite e bronchite. Inoltre, hanno una natura ricorrente e appaiono nei pazienti sin dalle prime settimane di vita.
Lo sviluppo dell'infezione nei polmoni porta ad un ulteriore aumento del contenuto mucoso nei bronchi, nonché ad un aumento della viscosità dell'espettorato, che comporta un rischio significativo per il paziente, specialmente nei primi anni di vita. In casi particolarmente difficili, può essere diagnosticato insufficienza respiratoria che spesso porta alla morte.
Per capire meglio la condizione in cui si trovano i pazienti affetti da fibrosi cistica, immagina di essere sempre in una maschera antigas. Allo stesso tempo, il filtro di aspirazione di questo dispositivo diventa sempre più intasato e gradualmente passa una minore quantità di ossigeno necessaria al corpo per respirare.
La stessa cosa accade con il considerato malattia genetica. Nella maggior parte dei pazienti, i polmoni svolgono le loro funzioni non più di un quarto.
Forme di fibrosi cistica
Esternamente, la malattia può manifestarsi sotto forma di una varietà di sintomi e segni. Dipende dalla natura e dalla gravità dei cambiamenti negativi nel corpo, dalla presenza o dall'assenza di qualsiasi complicazione, così come dall'età del paziente.
Basta distinguere diverse forme di patologia:
- polmonare (respiratorio o bronchiale);
- E .;
- misto (colpisce sia i polmoni che il tratto gastrointestinale);
- ostruzione del meconio del retto;
- atipico;
- cancellati.
Va notato che la classificazione di cui sopra è solo approssimativa e indicativa. Ciò è dovuto al fatto che la fibrosi cistica, che colpisce alcuni organi, ha un effetto negativo sul corretto funzionamento degli altri. Ad esempio, il blocco del muco dei canali bronchiali nei polmoni può essere accompagnato da danno intestinale e patologia in organi digestivi parallelo al sistema respiratorio.
Ogni singolo caso di diagnosi della fibrosi cistica può avere un quadro clinico differente. Ma il più delle volte i seguenti organi soffrono:
- intestini;
- luce;
- pancreas;
- il fegato.
In questo caso, in nessun caso c'è stato un effetto negativo della fibrosi cistica sulle capacità mentali del paziente.
Una forma atipica o lenta di un disturbo genetico viene solitamente diagnosticata per caso durante gli esami medici di routine. Ad esempio, in età adulta, è possibile stabilire segni di micoviscia durante l'esame del sistema riproduttivo per determinare le cause dell'infertilità maschile. In questo caso, l'incapacità di avere figli è solo una violazione concomitante. E la ragione principale dovrebbe essere considerata proprio la fibrosi cistica.
Questa malattia genetica negli uomini causa la cosiddetta azoospermia, cioè la completa assenza di cellule germinali maschili nel liquido seminale secreto. La fibrosi cistica, come è emerso durante la ricerca medica, colpisce il cordone spermatico, riducendone le dimensioni e danneggiando i tessuti circostanti.
A proposito, a volte i problemi con la funzione sessuale possono essere presenti in pazienti che sono solo portatori del gene mutato.
Nelle donne in età riproduttiva, la patologia provoca una diminuzione della capacità di dare alla luce bambini, che è causata da un aumento della viscosità del muco nel canale cervicale della cervice. Questo impedisce agli spermatozoi di entrare nella cavità dell'organo riproduttivo principale di una donna e di fecondare l'uovo lì.
I sintomi della fibrosi cistica polmonare
Consideriamo più in dettaglio la forma respiratoria della patologia.
Inizialmente, un paziente con un gene della fibrosi cistica presente sta vivendo debolezza generale e apatia, e la pelle diventa pallida. Il paziente inizia a perdere peso in maniera drastica, nonostante la grande quantità di cibo consumato.
Con una forma grave della malattia, già dai primi giorni dopo la nascita, una piccola tosse inizia a tormentare una persona, il cui carattere si intensifica nel tempo. Questi sintomi nelle loro manifestazioni esterne assomigliano a pertosse.
Poi viene l'espettorato, che ha una consistenza molto spessa. Nel caso di microflora patogena aggiuntiva (per esempio, stafilococco, menzionata nelle sezioni precedenti), il muco diventa purulento.
Le secrezioni intrapolmonarie secrete dai bronchi, acquisendo una consistenza più viscosa della necessaria, diventano la causa del blocco delle vie respiratorie. Nei casi più gravi è possibile far collassare qualsiasi parte del polmone, cioè l'emergere della cosiddetta atelettasia.
Il decorso della fibrosi cistica polmonare nei neonati può essere caratterizzato da danni alle sacche aeree alveolari, che forniscono uno scambio di ossigeno e di molecole di anidride carbonica tra i polmoni e l'aria.
Polmonite con fibrosi cistica
La malattia spesso porta allo sviluppo della forma più grave di polmonite, durante la quale si verificano spesso infiammazioni purulente del tessuto polmonare interno e formazione di cavità purulente in questo organo. La polmonite causata dalla fibrosi cistica è solo bilaterale.
Un certo numero di pazienti a volte mostra segni di tossicosi. Questo stato di malattia è causato dall'esposizione agli organi interni di una persona di varie tossine e altri fattori esterni. Il quadro clinico considerato in alcuni casi conduce allo shock del paziente.
La fibrosi cistica polmonare è accompagnata da violazioni dei seguenti sistemi corporei:
- nervoso;
- respirazione;
- circolazione del sangue;
- processi metabolici.
La polmonite derivante dal disturbo genetico descritto diventa cronica e si trasforma in cosiddetta pneumosclerosi. Con questo termine si intende un processo patologico, durante il quale si verifica una crescita anormale del tessuto connettivo nei polmoni, che causa una perdita di elasticità nello strato epiteliale e un disturbo del processo di scambio di gas.
Oltre alla pneumosclerosi, è possibile diagnosticare le bronchiectasie, cioè un aumento delle dimensioni dei bronchi, accompagnato da un cambiamento nella loro struttura cellulare e ispessimento delle pareti.
Occasionalmente, c'è una sindrome del "cuore polmonare", cioè un aumento del lato destro del cuore a causa di un aumento della pressione sanguigna in piccolo circolo di circolazione sanguigna che è stato causato da polmonite prolungata e patologie che accompagnano questo processo.
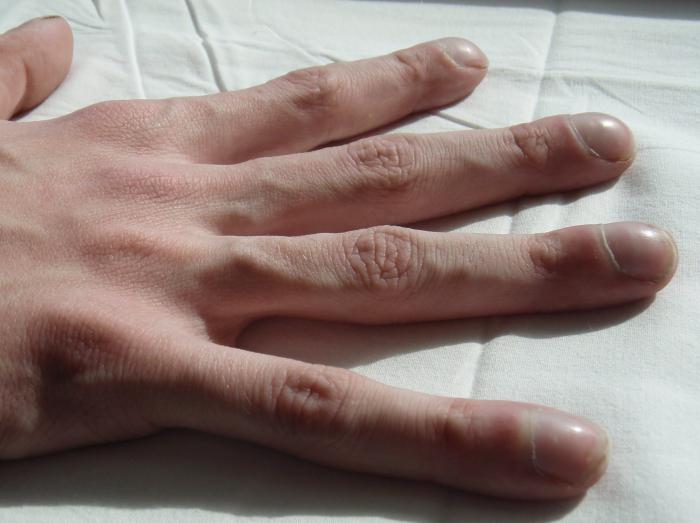
I sintomi esterni della polmonite da fibrosi cistica sono i seguenti. La pelle del paziente diventa marrone. Le membrane mucose visibili del corpo e la punta delle dita sugli arti superiori e inferiori acquisiscono una tinta bluastra.
Il paziente soffre di mancanza di respiro, pur essendo a riposo, il torace diventa una botte. Le ultime falangi delle dita si espandono in modo caratteristico, il che indica chiaramente la fibrosi cistica. Le foto delle cosiddette "dita di Ippocrate" possono essere trovate nei libri di consultazione medica. Una persona inizia a muoversi un po ', mentre il suo peso corporeo scende rapidamente, nonostante la grande quantità di cibo consumato.
I sintomi della fibrosi cistica intestinale
La malattia causa una violazione del corretto funzionamento del tratto gastrointestinale del corpo umano a causa del malfunzionamento delle ghiandole endocrine.
Molto spesso, questa forma di fibrosi cistica viene diagnosticata quando un neonato viene trasferito alla nutrizione artificiale parziale o completa. La disfunzione dello stomaco e dell'intestino porta al fatto che il cibo che entra nel corpo non si scompone nelle sue parti costituenti (proteine, grassi, carboidrati) e, di conseguenza, non vengono assorbiti nel sistema circolatorio.
La presenza nell'intestino di una grande quantità di cibo indigesto contribuisce ai processi di decomposizione. Questo, a sua volta, causa la formazione di tossine:
- idrogeno solforato;
- ammoniaca;
- ammine primarie e secondarie.
Inoltre, a causa dell'accumulo nel tratto gastrointestinale di una grande quantità di gas interni, molti pazienti con fibrosi cistica si lamentano di gonfiore.
I processi negativi descritti richiedono visite più frequenti al bagno. A volte può anche andare alle "poly-feces", cioè alle visite anormalmente frequenti in bagno per la defecazione. A volte questo bisogno si pone in 4, e anche 8 volte più spesso che in una persona normale sana.
La patologia considerata può portare al prolasso del retto. Questo disturbo si verifica in circa il 20% dei pazienti con fibrosi cistica intestinale.
trattamento
Attualmente non esiste una cura per i sintomi della fibrosi cistica. Di solito, i medici usano la cosiddetta terapia sintomatica.
Ad esempio, i farmaci possono essere utilizzati per ripristinare il corretto funzionamento del tratto gastrointestinale o del sistema respiratorio. In casi particolarmente difficili, vengono applicate misure terapeutiche più gravi.

Disturbi negli organi digestivi richiedono al paziente di mantenere una dieta, che aiuta a prevenire il prolasso rettale. Inoltre, dovresti monitorare attentamente il giusto equilibrio nelle proteine, nei grassi e nei carboidrati alimentari.
Va notato che la diagnosi tempestiva è la chiave per il successo del trattamento dei sintomi della fibrosi cistica. Altrimenti, la possibilità di morte. Soprattutto per i pazienti la cui età non ha raggiunto 1 anno.
conclusione
La fibrosi cistica è una malattia genetica che si verifica in un neonato solo quando quest'ultimo ha ricevuto due geni mutati dai suoi genitori.
Questa patologia provoca processi negativi in molti organi e richiede costante attenzione e varie misure volte a trattare i sintomi emergenti. Ad esempio, la forma polmonare della fibrosi cistica richiede una terapia farmacologica mirata a combattere la polmonite risultante. E l'aspetto del tratto gastrointestinale - dieta e controllo della microflora permanente.
Altrimenti, l'insorgenza di complicazioni molto gravi, e talvolta la morte.
Non dovresti trascurare la tua salute.